Read mapping
Splicing is a crucial process in gene expression where introns (non-coding regions) are removed from pre-mRNA transcripts, and exons (coding regions) are joined together.
Credit: Cold Spring Harbor Laboratory
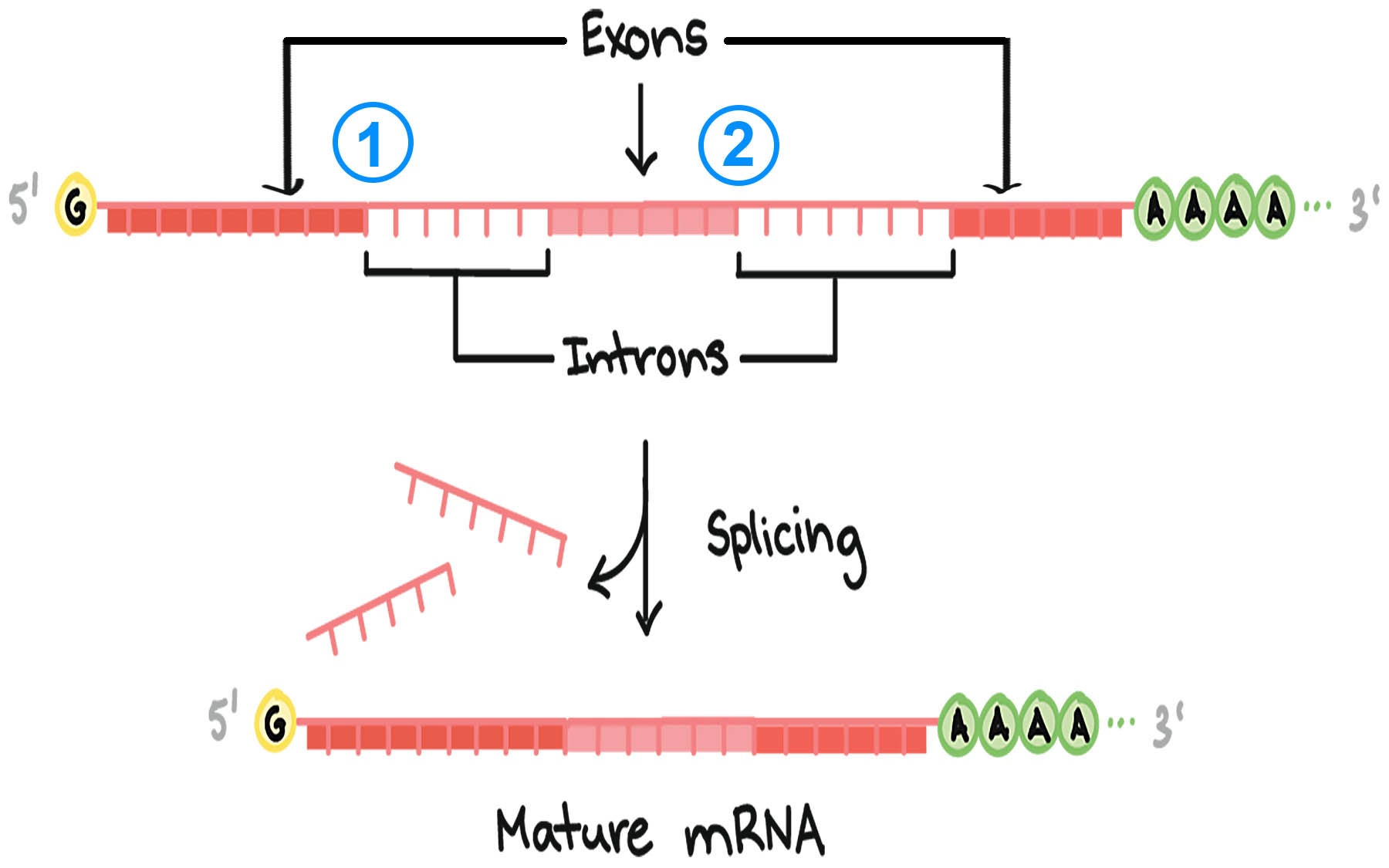
This process allows a single gene to produce multiple proteins through alternative splicing, significantly contributing to transcript diversity and the complexity of proteomes in eukaryotic organisms. By selectively including or excluding RNA segments, cells can generate various proteins from a limited number of genes, playing a fundamental role in cellular functions and organism development.
The necessity of splice-aware alignment tools for RNA-seq (RNA sequencing) data analysis stems from the challenge of accurately mapping short RNA-seq reads to a reference genome. These reads may span exon-exon junctions, where the genetic sequence is discontinuous due to the removal of introns during splicing. Traditional alignment tools, designed for contiguous genomic sequences, may fail to correctly align reads across exon-exon junctions, leading to inaccurate gene expression quantification and misunderstanding of the transcriptome’s complexity. Splice-aware aligners are designed to handle these complexities by identifying potential splicing events and mapping reads spanning exon-exon junctions. This enables precise quantification of gene expression levels, identification of alternative splicing events, and a deeper understanding of the transcriptome’s dynamic nature, essential for studies on gene regulation, disease mechanisms, and therapeutic strategies.
Challenges
Splice junctions
The primary challenge in splice-aware alignment is the identification of splice junctions. Unlike other genomic regions, exon-exon junctions (the points where two exons are joined together after introns are removed) are not contiguous in the genome. This non-contiguity makes it challenging to align reads that span these junctions because the alignment tools must infer the splicing events that connect the exons. This inference process is computationally intensive due to:
- There is a vast diversity of potential splicing events, with thousands of possible combinations in which exons can be joined.
- There is a need for high accuracy in distinguishing true splice junctions from sequencing errors or variations.
- The computational complexity of aligning short reads to a reference genome, considering the potential for multiple splicing options.
Several computational tools and algorithms have been developed to tackle the challenge of identifying splice junctions. These tools employ strategies such as using known splice junctions from databases, predicting junctions based on sequence motifs, or employing machine learning techniques to infer splicing events from RNA-seq data.
Alternative splicing
The vast potential for alternative splicing in eukaryotic organisms means that there can be many possible transcripts for a given gene, each potentially producing different protein variants. Accurately mapping reads to these variants requires sophisticated computational approaches that can handle the ambiguity and complexity of the transcriptome.
Genomic Variability and Errors
Complicating matters further, genomic variability among individuals and sequencing errors can mimic splicing events or obscure real splice junctions, making the task of accurately identifying and mapping reads to the correct genomic locations even more challenging.
Short reads
Short reads are small sequences of DNA, typically ranging from 50 to 300 base pairs in length, produced by NGS technologies. Despite their critical role in genomic analysis, these short sequences pose significant challenges for accurate genome alignment. Due to their limited length, short reads may not contain enough unique information to unambiguously map them back to their original location in the genome. This is akin to finding the exact location of a specific sentence in a large book by only having a few words from that sentence.