Scoring
In DNA sequence alignment, scoring is crucial for quantifying the similarity or difference between two DNA sequences. This method helps identify the best possible alignment between sequences, revealing evolutionary relationships, gene function, and genetic variations. The scoring concept for nucleotide substitutions, insertions, deletions, and gaps is designed to reflect their biological significance.
Substitutions
A nucleotide substitution occurs when one nucleotide (A, T, C, or G) is replaced by another in the sequence.
Credit: Pathwayz
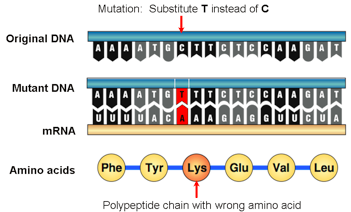
Scoring typically involves assigning positive scores to matches (identical nucleotides at the same position in both sequences) and negative scores to mismatches (different nucleotides at the same position). For example, a scoring system might use +1 for a match and -1 for a mismatch. This simple scheme rewards alignments of more similar sequences and penalizes those that differ.
Some scoring systems differentiate between
- transitions: substitutions between two purines (A ↔ G) or two pyrimidines (C ↔ T)
- transversions: substitutions between a purine and a pyrimidine.
Since transitions occur more frequently in evolution than transversions, some scoring schemes might penalize them less severely.
Insertions or deletions
These events represent the insertion or deletion (also called indel) of nucleotides in a sequence relative to another. Scoring for indels involves introducing gaps in the alignment to maintain optimal sequence alignment. The scoring system usually penalizes such gaps to reflect the biological cost of indels.
Credit: Hackbright Academy

String distances
Hamming
The Hamming distance between two strings of equal length is defined as the number of positions at which the corresponding symbols differ. Imagine you have two strings: GATTACA and GACTATA. The Hamming distance between these two strings would be two because there are two positions at which the nucleotides differ. This measure quantifies the minimum number of substitution operations needed to change one string into another.
The primary limitation of the Hamming distance is its applicability only to sequences of the same length. This is because the Hamming distance measures the difference at specific positions between two sequences. If the sequences are of different lengths, the concept of position-wise comparison doesn’t hold, making the Hamming distance calculation infeasible. Therefore, it cannot account for insertions or deletions common in DNA sequences and text strings, limiting its use in cases where these variations are significant.
Levenshtein
The Levenshtein distance, often called the edit distance, measures the minimum number of single-character edits required to transform one sequence into another. These edits can include insertions, deletions, or substitutions of characters. A key characteristic that sets the Levenshtein distance apart from other metrics like the Hamming distance is its ability to handle sequences of different lengths, making it incredibly versatile for comparing any two strings.
For example, to transform the word “kitten” into “sitting,” the Levenshtein distance would be 3:
- Substitute “k” with “s”
- Substitute “e” with “i”
- Insert “g” at the end
This metric provides a clear, numerical value representing the “distance” or difference between two sequences.
Unlike metrics limited to comparisons of equal-length sequences, the Levenshtein distance accommodates differences in sequence length, broadening its applicability. By measuring insertions, deletions, and substitutions, the Levenshtein distance captures the full spectrum of possible edits, providing a comprehensive view of the similarity or dissimilarity between sequences.